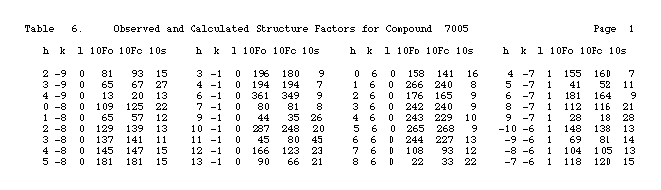
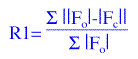Once a structure solution has been achieved, either by Heavy
Atom Methods or Direct Methods, there are actually two structure models, a calculated model based on
the approximate co-ordinates obtained from interpretation of a three dimensional electron density map: